Hey, great job, everyone! The poster session last Friday was a great success. It was neat to meet so many of your parents, PI's, and mentors. I'm so glad to hear that so many of you enjoyed your summer research experiences.
Thanks so much to all of those who kept posting to the blog throughout the summer. I think it was an overall success, and we'll be adding it as a requirement to next year's program.
Once again, great job everyone! Please keep us updated about your career goals and where you end up for college! Good luck this next year and enjoy the rest of the summer!
Tuesday, July 31, 2007
Thursday, July 26, 2007
Going out with a bang!
I can't believe its already the very last day working in the labs. It'll be sad to leave the lab but I'm excited for the poster session tomorrow!
Today was transplant day for one of the two patients waiting right now--very exciting!!
I was able to observe in the operating room with Dr. Markert, Julie (who works in the lab) and two other students (Dave and Ashley). After we carried over all the slices of thymus in a plastic bin and suited up in bunny-suits, hair and shoe covers, and masks we entered the operating room. A resident was actually performing the surgery with one or two doctors and several nurses, and us. It was a pretty full house but luckily we were in a pretty big room. They had started to put the central line into the baby when we arrived and it took them a while to get it positioned correctly. They start by putting a needle near the neck and then using it to guide a wire through a vein all the way to the heart, and then thread a tube over the wire and pull the wire out. They used x-rays to watch the position on the wire which we could watch on a screen. The central line will be used to give the patient calcium and nutrients while he's waiting for stomach surgery. They then started the transplant procedure by making an incision on both thigh muscles. Fortunately, one of the students was able to recognize at this point that they were not meant to be a surgeon and had to go sit down right away. I went out into the hall when the person fainted. Luckily, we were already in a hospital and some nice doctor brought us a wheelchair and some food. And so...i missed most of the actual transplant surgery. I was able to go back in the operating room just in time to see the last thymus slice inserted and watched them sew it up. Needless to say, it was a very interesting day at the hospital all around and a high light of the summer!
I'm so grateful for this opportunity, i can't imagine a better way to have spent time working this summer. Everyone in the lab made it such an enjoyable and amazing experience. Thanks to everyone in the group too, it's been fun!
Today was transplant day for one of the two patients waiting right now--very exciting!!
I was able to observe in the operating room with Dr. Markert, Julie (who works in the lab) and two other students (Dave and Ashley). After we carried over all the slices of thymus in a plastic bin and suited up in bunny-suits, hair and shoe covers, and masks we entered the operating room. A resident was actually performing the surgery with one or two doctors and several nurses, and us. It was a pretty full house but luckily we were in a pretty big room. They had started to put the central line into the baby when we arrived and it took them a while to get it positioned correctly. They start by putting a needle near the neck and then using it to guide a wire through a vein all the way to the heart, and then thread a tube over the wire and pull the wire out. They used x-rays to watch the position on the wire which we could watch on a screen. The central line will be used to give the patient calcium and nutrients while he's waiting for stomach surgery. They then started the transplant procedure by making an incision on both thigh muscles. Fortunately, one of the students was able to recognize at this point that they were not meant to be a surgeon and had to go sit down right away. I went out into the hall when the person fainted. Luckily, we were already in a hospital and some nice doctor brought us a wheelchair and some food. And so...i missed most of the actual transplant surgery. I was able to go back in the operating room just in time to see the last thymus slice inserted and watched them sew it up. Needless to say, it was a very interesting day at the hospital all around and a high light of the summer!
I'm so grateful for this opportunity, i can't imagine a better way to have spent time working this summer. Everyone in the lab made it such an enjoyable and amazing experience. Thanks to everyone in the group too, it's been fun!
Wednesday, July 25, 2007
the end, oh sorrow
Oh no! Howard Hughes is coming to an end...and it coincides with the last harry potter book too! my life is going to be slightly depressing after this. i had so much fun with everyone, and yesterday's dinner and hogwarts sculpting session and outside photo-attackingness were aMaZiNg. i can't wait for friday's poster session; we should all be really proud of our work! More later, i think i'm going to try to sneak my way into a gastric fluid extraction show.
~love from linda
~love from linda
Monday, July 23, 2007
The End
Well guys, this is the end. The final chapter of our time here. It's been a great experience, and I know everybody's excited/anxious about presenting their projects. Personally, I am not quite finished with mine. But I still have one more day to tie up loose ends. I have my conclusion written, but not my results. I'm sure I'll finish it up tonight. I can't believe all that I've learned these past 6 weeks- sequencing, PCR, gels, purifications, specs, bioinformatics, statistical tests, sea urchin development, and the ridiculously high cost of biological research. Now I know what grants are for. Though I'm still not sure what I want to do with my life career wise, I think I've learned a lot about biology and lab research this summer. There is a huge difference between just reading about something and actually doing it. Thanks to everyone in my lab and to all the ladies with the Howard Hughes Program- you guys have been really great! Thanks to all the speakers and thanks to the sea urchins for sacrificing your little lives for the greater good! So long.
Results
The second to the last week was surprisingly exciting. I was thrilled to see if we could find any associations using the lab data. Dr. Allison and Melanie assisted me in using statistical approaches to analyze my data. One of the interesting things that we found was that child IQ was predicted by genotype at one VDR SNP (RS7975128, p=0.01), and maternal smoking within the child’s first 7 years of life (p=0.03). However, the interaction between the VDR SNP and maternal smoking was not significant (p=0.40). The A/G and G/G genotypes of RS7975128 were associated with lower IQ, as was the occurrence of maternal smoking. I also attended a lab meeting and a clinical meeting.
Thursday, July 19, 2007
Formatting
So this week I was supposed to work on analyzing the data I had gathered. I had hundreds of sequences available, but not all of the genes had enough individuals to be useful. I deleted all the genes that did not have seven or more individuals available. With less than 14 haplotypes, you can’t make very reliable conclusions about that particular gene. So it turned out that only ten out of twenty-seven genes were fit for analyzing. First, I had to transfer the sequencher files to MacClade, a program that helps you align the base pairs so that you aren’t starting in the middle of a codon. The MacClade program lets me see all the sequences in the gene at the same time. The SNPs show up very clearly because each base is a different color, so if there were no SNPs, the gene sequences would just look like pinstripes. So then I had to export the MacClade files as fasta files. I had to format all the sequences individually, which was time consuming, but now I am finally done and all I have to do is give Ralph, who has written programming to analyze selection and variation frequency for sequences. I was formerly using a program called DNAsp, which essentially does the thing that Ralph’s program does, but it would not read any of the data files I made on my computer. Also, Ralph’s program not only gives you the numbers, but also tells you how certain it is in this number. DNAsp just gives you the number. On Friday and Monday I’ll probably work on fixing my poster.
Sunday, July 15, 2007
Killifish Eggs and Hatchlings
Everything has been busy in the Eco-toxicology Lab getting ready for next week. We've have been dosing the fish eggs with various contaminants trying to find the LC 50 (the point at which half of the eggs are dead). When we have the LC 50 for fish from our control site, we can use that data to compare it to how fish from a contaminated site, the Elizabeth River, deal with the same amount of contamination.
This has been harder then we first expected. The first contaminant we exposed them to was Ammonium Chloride. We tried three dose responses with killifish eggs. In the first experiment, we tried doses ranging from 7.5 mg/L to 150 mg/L. The fish had no reaction and continued with development as usual. We tried another dose ranging from 250 mg/L to 1500 mg/L and once again the killifish eggs showed no response. We did a final, more extreme experiment with doses ranging from 2,500 mg/L to 10,000 mg/L. When the eggs showed no response to this as well, we came to the conclusion that the killifish are protected inside their eggs and that the ammonia does not penetrate the egg. We later did experiments in killifish hatchlings which did show a response to the ammonia. I never thought I'd be happy to say that something died, but after watching many fish eggs survive the ammonia doses, I was glad to deliver the news that hatchlings did show the response we expected to the ammonia and that we would be able to continue on with our research.
Although, this presented an obstacle in continuing with my research of whether Elizabeth River killifish have a fitness cost associated with their adaptation to PAH's. We could no longer do research in eggs because the pesticides we were exposing them to proved to be similar to the ammonia in the sense that the killifish eggs showed no response. Instead we would need to do our research with hatchlings. Killifish eggs take two weeks to hatch. A large quantity of them, 350, will be ready from hatching tomorrow but this set us back a bit in data collection. We will conduct the majority of our research on these hatchlings next week using the data that we have gathered in the last few weeks with the dose response experiments, to know what the correct doses are to reach the LC 50. It will be another busy week, ending of course with the release of the final Harry Potter book. I know many of us in the program are looking foreward to that.
This has been harder then we first expected. The first contaminant we exposed them to was Ammonium Chloride. We tried three dose responses with killifish eggs. In the first experiment, we tried doses ranging from 7.5 mg/L to 150 mg/L. The fish had no reaction and continued with development as usual. We tried another dose ranging from 250 mg/L to 1500 mg/L and once again the killifish eggs showed no response. We did a final, more extreme experiment with doses ranging from 2,500 mg/L to 10,000 mg/L. When the eggs showed no response to this as well, we came to the conclusion that the killifish are protected inside their eggs and that the ammonia does not penetrate the egg. We later did experiments in killifish hatchlings which did show a response to the ammonia. I never thought I'd be happy to say that something died, but after watching many fish eggs survive the ammonia doses, I was glad to deliver the news that hatchlings did show the response we expected to the ammonia and that we would be able to continue on with our research.
Although, this presented an obstacle in continuing with my research of whether Elizabeth River killifish have a fitness cost associated with their adaptation to PAH's. We could no longer do research in eggs because the pesticides we were exposing them to proved to be similar to the ammonia in the sense that the killifish eggs showed no response. Instead we would need to do our research with hatchlings. Killifish eggs take two weeks to hatch. A large quantity of them, 350, will be ready from hatching tomorrow but this set us back a bit in data collection. We will conduct the majority of our research on these hatchlings next week using the data that we have gathered in the last few weeks with the dose response experiments, to know what the correct doses are to reach the LC 50. It will be another busy week, ending of course with the release of the final Harry Potter book. I know many of us in the program are looking foreward to that.
BuSy Week With the ADHD samples!!!!
On Monday, I was able to plate my ADHD DNA using the Biomek robot. This made me extremely excited because I was finally able to start on my project. I left the plates to dry overnight at room temperature. The following day, I began working on my assays involving the ALAD gene and the VDR gene. I made 18 Taqman plates for 9 assays and ran 14 of those plates onto the PCR machine. I then scanned them on the 7900HT machine. Most of my TAqman went well except for two plates. Next, for Wednesday, I had to re-run the two plates that failed. I also ran 6 more plates on the PCR machine and made 2 plates for a single assay. I scanned those plates as well and they turned out great. For the rest of the day, I had to plate 20 extra plates using the Biomek robot. On Thursday, I made 5 Taqman plates in the morning. I ran them using the PCR machine and scanned them afterwards. I then added markers and detectors. Fortunately, they looked great. In the afternoon, I ran about 5 more plates and scanned them. In addition, the fire alarm went off again for the second time since I have been in CHG. On Friday, I really enjoyed the talk by Ms. Cleaver. It gave me many tips in applying for college. After that, I went to my lab and ran and scanned some plates that I had made prior. I also helped Kaia by making two agarose gels for her and running her PCR samples. Some looked good but the rest failed. Chris then helped me submit my data so that I could view them with a statistician next week. Unfortunately, two failed to submit but I’ll be able to do that on Monday. Overall, I had an exciting and this week was probably the busiest for me by far.
Sequencher
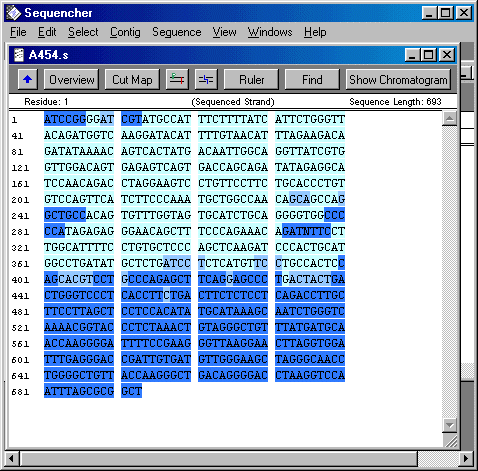
So this last week I focused on organising and cleaning up sequences in a Sequencher program. I had to find SNPs and label them by hand. Since I was reviewing 20-ish different 200 to 1000 bp sequences for 2 to 24 individuals each. THat's a lot of letters. So I didn't quite finish, but I hope to finish up on Monday. Not only did I label SNPs, I also cut off the ends of each sequence that were not reliable. Sequencher actually reads the chromatogram and translates the flourescent waves into four letters, the letters that encompass the genetic code (C, A, T, and G). Sometimes, though, Sequencher makes a bad call. When it miscalls a base or is not sure about a base, I have to fix that by changing the sequence. Once I have cleaned up all the sequences so that all sequences of the same primer pairs are the exact same length, then I will get help from some other people in the lab on analyzing the data. The picture is what the sequence looks like on sequencher. Dark blue bases mean that Sequencher is less sure about the base call, whereas light blue bases mean that Sequencher is pretty certain that the base is as it calls it.
Hi everyone!
For the last two weeks, I have been continuing to work on my Toluene experiments. Turns out that the results were not as promising as we had hoped since the E. coli conjugates do not seem to be breaking down the Toluene. :-( I have been testing different conditions to see if the toluene will degrade in more stressful environments, and some seem to be working a little better than the others. One possibility is that the original stock solution has been contaminated, since it has been sitting in the refridgerator for a while now. We should probably make a new stock solution on Monday. Hopefully Ruoting and I will be able to find advantageous conditions for the E. coli soon. :-D
Annie
Harry Potter and the Deathly Hallows
5 days. 4 hours. 27 minutes. 07 seconds.
For the last two weeks, I have been continuing to work on my Toluene experiments. Turns out that the results were not as promising as we had hoped since the E. coli conjugates do not seem to be breaking down the Toluene. :-( I have been testing different conditions to see if the toluene will degrade in more stressful environments, and some seem to be working a little better than the others. One possibility is that the original stock solution has been contaminated, since it has been sitting in the refridgerator for a while now. We should probably make a new stock solution on Monday. Hopefully Ruoting and I will be able to find advantageous conditions for the E. coli soon. :-D
Annie
Harry Potter and the Deathly Hallows
5 days. 4 hours. 27 minutes. 07 seconds.
Tuesday, July 10, 2007
Hey guys, it's Linda. Last week after talking to my PI, I found out that both of my experiments, 317 colony diameter growth and small vs. large colony bacteria biofilm growth seem to be working. : ) ! Additionally, in the former experiment, bacteria growth in a particular Fufu dilution spawned atypically large colonies, something that Bill said he'd never seen. So I shall now be taking on a mini-third experiment by replating some suspicious looking colonies to see if their proceeding generations will retain the same diameters.
The only slight problem this poses is that I will now have a more difficult time in trying to choose which project to focus on for my poster presentation. Additionally, I am worried that I will be short on time and will have a harder time acquiring all the data when my time is split three-ways. In eight minutes now, I'll be rock-climbing with some people from our lab for the first time ever. I hope to survive so I can at least finish the last book of the Harry Potter series. I'll show you my scrapes and cracked jaw bone tomorrow. Chow!
The only slight problem this poses is that I will now have a more difficult time in trying to choose which project to focus on for my poster presentation. Additionally, I am worried that I will be short on time and will have a harder time acquiring all the data when my time is split three-ways. In eight minutes now, I'll be rock-climbing with some people from our lab for the first time ever. I hope to survive so I can at least finish the last book of the Harry Potter series. I'll show you my scrapes and cracked jaw bone tomorrow. Chow!
Monday, July 9, 2007
Making a Slide
 So. Here's how I make a slide.
So. Here's how I make a slide.1. Line up embryos with a probe (pointy thing).

2. Put them on a slide (see the little white smudge in the middle?).
We put them in halocarbon oil so they can still breathe!

3. Put the slide in the microscope.

4. Turn on the laser. (488nm = blue)

5. Viola! (10x lens)
This is basically what I do for my experiment. Today, I did a time lapse series. I took pictures every hour to see how GFP expression varies over time. Hopefully, I'll be able to fit a line or a curve to my data points tomorrow.
Drosophila Drosophila Drosophila (try saying it 3 times fast)
Hi! Sorry for the long absence, it's been pretty busy. Anyway, in the last few weeks my project has really developed. As I mentioned before, I will be measuring the fluorescence levels of several different GFP fly lines. My lab will use this information to choose bright or dim embryos depending on the needs of their experiment (ex: you have a red fluorescent protein you want to image with the green at the same time. the red is dim, so you want to choose a dim green as well so the faint red is not drowned out).
Because my project compares different pictures taken at different times, we needed to figure out a way to make a "control" in each picture. The pictures are different because the embryos fluoresce under a 488nm laser. That laser can vary in power and make 2 different fluorescing embryos look the same. Different embryos also require different laser powers to create 'optimal' pictures--not too bright or too dim.
The first thing we tried was fluorescent beads. The idea made sense: squirt some of these 6um beads (embryos are about 150 micrometers by 400um) onto each slide. Take pictures. Use the ratios of bead brightness from each picture to compare embryo brightnesses (ex: in one picture, the beads have a pixel value of 1. in the other, 2. to compare, double the brightness of the embryo in picture one).
But in practice, this was not so easy. First of all, the beads needed to be kept in the dark and at around 4C all the time. This meant I had to do all the squirting in the dark in the 'cold room' where my lab works with proteins. Needless to say, it was pretty hard. I would either miss the spot on the coverslip (if the embryos and beads weren't in the same frame for pictures, it wouldn't work), the oil I was injecting into would freeze (you put embryos in halocarbon oil to keep them alive under a coverslip), or if I did get the beads right, they would mess themselves up. Once, the computer measured the 3% beads as WAY brighter than the 30%. So beads weren't so helpful. (one of the grad students had ordered them a long time ago to do what I am now, but got frustrated and decided it wasn't worth the time)
But luckily we found a paper. And it described a method that did not include beads. Our new method allows us to adjust laser power for each embryo and then normalize the values to make them comparable. Basically, you view the results as if you had taken each picture at 1% laser power. First, you raise the laser power to make the embryo you are viewing a certain brightness. You take the picture, take the mean brightness of the embryo (with a histogram on the computer) and divide by the laser power you used (ex: mean brightness=100 laser power=25% normalized brightness=4).
I'm really enjoying the new method we found for two reasons: 1) I get to use the really fancy 510 confocal microscope (I will post pictures) 2) I feel like I got to have creative/intellectual input when it came to how we were going to implement it. No one in the lab has done it before, so we kind of got to start from scratch.
Anyway, now I'm going to put up some pictures from a few weeks ago. Next time: my side projects-- inverse PCR and gene mapping!
Because my project compares different pictures taken at different times, we needed to figure out a way to make a "control" in each picture. The pictures are different because the embryos fluoresce under a 488nm laser. That laser can vary in power and make 2 different fluorescing embryos look the same. Different embryos also require different laser powers to create 'optimal' pictures--not too bright or too dim.
The first thing we tried was fluorescent beads. The idea made sense: squirt some of these 6um beads (embryos are about 150 micrometers by 400um) onto each slide. Take pictures. Use the ratios of bead brightness from each picture to compare embryo brightnesses (ex: in one picture, the beads have a pixel value of 1. in the other, 2. to compare, double the brightness of the embryo in picture one).
But in practice, this was not so easy. First of all, the beads needed to be kept in the dark and at around 4C all the time. This meant I had to do all the squirting in the dark in the 'cold room' where my lab works with proteins. Needless to say, it was pretty hard. I would either miss the spot on the coverslip (if the embryos and beads weren't in the same frame for pictures, it wouldn't work), the oil I was injecting into would freeze (you put embryos in halocarbon oil to keep them alive under a coverslip), or if I did get the beads right, they would mess themselves up. Once, the computer measured the 3% beads as WAY brighter than the 30%. So beads weren't so helpful. (one of the grad students had ordered them a long time ago to do what I am now, but got frustrated and decided it wasn't worth the time)
But luckily we found a paper. And it described a method that did not include beads. Our new method allows us to adjust laser power for each embryo and then normalize the values to make them comparable. Basically, you view the results as if you had taken each picture at 1% laser power. First, you raise the laser power to make the embryo you are viewing a certain brightness. You take the picture, take the mean brightness of the embryo (with a histogram on the computer) and divide by the laser power you used (ex: mean brightness=100 laser power=25% normalized brightness=4).
I'm really enjoying the new method we found for two reasons: 1) I get to use the really fancy 510 confocal microscope (I will post pictures) 2) I feel like I got to have creative/intellectual input when it came to how we were going to implement it. No one in the lab has done it before, so we kind of got to start from scratch.
Anyway, now I'm going to put up some pictures from a few weeks ago. Next time: my side projects-- inverse PCR and gene mapping!
FAmily Visit
I was able to observe my first family visits on a Friday. So far, that was probably one of the highlights of my experience with the program. I watched as the entire family of four completed a series of behavioral and psychological tests as well as developmental histories to see if they could be diagnosed with ADHD. There are three rooms in the clinic area: a computer room, an interview room, and a blood room. One member of the family goes into the computer and interview room at a time. In the computer room, letters appear on the computer simultaneously at varying time intervals and every time a letter, other than “X”, appears, the participant has to push the space bar. This test takes place for exactly fourteen minutes. After a while, one of the young participants that I watched became bored of the test, so he continuously and randomly just began pushing the space bar, increasing his impulsitivity score. The interview room is where the psychologist assesses them and gives them an IQ test by asking several questions as well as by allowing them to complete spatial and hands-on tasks. Finally, after everyone in the family completes these tests, there blood is drawn and set to the DNA bank. Unfortunately, one of the children refused to get a blood draw. Overall, the entire family visit lasted for a period of three hours and thirty minutes.
Last week was a pretty slow week with most of the group on vacation. I continued to watch the jurkat cells and count them on a pretty regular basis. This week there should be enough cells to harvest them and extract RNA. I also observed the extracting of PBMC's (peripheral blood mononuclear cells) which contain the lymphocytes and monocytes (cells of the immune system). When spun at high speeds the blood separates into different levels according to density. The lymphocytes and monocytes form a buffy coat under the layer of plasma that you pipette out. It's important to get good separation of the blood for clear results in many of the tests in the lab. Once the pbmc's were separated Jie showed me how they select the T cells (a type of lymphocyte) from the sample magnetically. T cells express CD3 on the cell surface. The white blood cells are mixed with CD3 antibodies that are attac
them on a pretty regular basis. This week there should be enough cells to harvest them and extract RNA. I also observed the extracting of PBMC's (peripheral blood mononuclear cells) which contain the lymphocytes and monocytes (cells of the immune system). When spun at high speeds the blood separates into different levels according to density. The lymphocytes and monocytes form a buffy coat under the layer of plasma that you pipette out. It's important to get good separation of the blood for clear results in many of the tests in the lab. Once the pbmc's were separated Jie showed me how they select the T cells (a type of lymphocyte) from the sample magnetically. T cells express CD3 on the cell surface. The white blood cells are mixed with CD3 antibodies that are attac hed to an iron molecule. These micro beads only attach to the cells expressing CD3 and the whole sample is run through a filter with a magnet. It was cool to see the process from beginning to end starting with whole blood and ending with a specific type of lymphocyte.
hed to an iron molecule. These micro beads only attach to the cells expressing CD3 and the whole sample is run through a filter with a magnet. It was cool to see the process from beginning to end starting with whole blood and ending with a specific type of lymphocyte.
them on a pretty regular basis. This week there should be enough cells to harvest them and extract RNA. I also observed the extracting of PBMC's (peripheral blood mononuclear cells) which contain the lymphocytes and monocytes (cells of the immune system). When spun at high speeds the blood separates into different levels according to density. The lymphocytes and monocytes form a buffy coat under the layer of plasma that you pipette out. It's important to get good separation of the blood for clear results in many of the tests in the lab. Once the pbmc's were separated Jie showed me how they select the T cells (a type of lymphocyte) from the sample magnetically. T cells express CD3 on the cell surface. The white blood cells are mixed with CD3 antibodies that are attached to an iron molecule. These micro beads only attach to the cells expressing CD3 and the whole sample is run through a filter with a magnet. It was cool to see the process from beginning to end starting with whole blood and ending with a specific type of lymphocyte. It also looks like I've got the rest of the data I need for my project. It's exciting to see the last bits come together. So far our data suggests that the CD56hi cells we were looking for are present pre-transplant in babies with no thymus function. This does not support the hypothesis in another paper, based on mice, which proposes that this type of cell is derived from the thymus.
I am continuing to learn more about the primitive number systems in human infants and lemurs as the summer progresses. In the baby lab, we have finished up the Ordinal Low-Cognitive Demand with 9 and 11 month olds, and are now moving on to different things (although I will still be using the Ordinal-LCD paradigm for my project). 11 month olds are now doing an Ordinal Transfer study, which shows sequences of stimuli that increase. They are habituated to these images, and then tested on the opposite condition (i.e. if they are habituated to ascending sequences they are tested on descending ones). We are beginning to set up two visit experiments with 9 month olds; on their first visit we show them a paradigm in which we know they succeed (where surface area, number and perimiter I believe all increase or decease), and then on their second we show them stimuli where only numerosities increase or decrease. We think that perhaps if we give these infants some sort of "practice" they may succeed.
The DLC is always fun, and this week will be a little different. We are "shaping" two new female mongoose lemurs (Guadalupe and Eduardo)-teaching them how to use the touch screen (contrary to what many believe, just because Rosalita may have watched Eduardo do the experiment, she still has no idea how to even approach the computer). Aristides will also start again with the Risk paradigm after being out sick for quite a while. Aristides will be so excited, as whenever we walk by with the machine he thinks it's his turn and is ready to go! Finally he'll be able to. Also thanks to Aristides (a ringtail) I have data to use for my project. Aristides has finished the experiment that Teres is now running (that I am using for my project).
Hope everyone had a great 4th and a weekend! This summer is going by just way too fast....
The DLC is always fun, and this week will be a little different. We are "shaping" two new female mongoose lemurs (Guadalupe and Eduardo)-teaching them how to use the touch screen (contrary to what many believe, just because Rosalita may have watched Eduardo do the experiment, she still has no idea how to even approach the computer). Aristides will also start again with the Risk paradigm after being out sick for quite a while. Aristides will be so excited, as whenever we walk by with the machine he thinks it's his turn and is ready to go! Finally he'll be able to. Also thanks to Aristides (a ringtail) I have data to use for my project. Aristides has finished the experiment that Teres is now running (that I am using for my project).
Hope everyone had a great 4th and a weekend! This summer is going by just way too fast....
ADHD
On Monday, I continued working on my methods section until Chris, my mentor, arrived. Surprisingly, one of my friends from school was assigned to the same lab as me and she had just begun working there. So, she had to observe Chris and I for that day. Prior to lunch, I worked on two PCR plates and four Taqman plates. Then after lunch, I had to make an agarose gel to test my PCR. My PCR worked; however, it was not that great since some of the wells looked as if they were missing DNA samples. I assisted my friend in practicing using the pipette. Then, I completed one more PCR plate and scanned my Taqman plates afterwards. Fortunately, all of my Taqman worked out well. For the rest of the time, I worked on my methods section again and completed my blog.
Tuesday, I worked on a Taqman plate and two PCR plates. After running them on the machine, I had to wait until they finished, so that they could be tested on an agarose gel. After lunch, I tested the samples on the gel. My results were better than my results on the previous day. So, I completed two more PCR plates using a different primer, and ran them on the PCR machine. Then Chris taught me a new method using Sephadex to filter the DNA from our PCR. It was fun being able to learn a new method. This was a gel like substance that allows the DNA to separate from the excess materials used during the PCR reaction. After that, I worked on my blog.
I had a wonderful Independence Day celebration with my friends on Wednesday and returned to work on Thursday. I felt so excited to receive my first paycheck. When I came to the lab, I made an agarose gel and ran my previous PCR samples on it. It actually worked out well. Afterwards, I made another PCR plate prior to lunch. I made another PCR plate after lunch and I also began labeling my ADHD plates. I then waited for my PCR plates to finish running so that they could be tested on an agarose gel.
Friday was a great day! I began my day by making a PCR plate. I then ran my PCR agarose gel that I made earlier in the week. Fortunately, all my samples worked. Next, I ate lunch early because I was invited to observe a family visit. This is when the proband as well as the entire family come into the clinic area to be tested for ADHD.
Tuesday, I worked on a Taqman plate and two PCR plates. After running them on the machine, I had to wait until they finished, so that they could be tested on an agarose gel. After lunch, I tested the samples on the gel. My results were better than my results on the previous day. So, I completed two more PCR plates using a different primer, and ran them on the PCR machine. Then Chris taught me a new method using Sephadex to filter the DNA from our PCR. It was fun being able to learn a new method. This was a gel like substance that allows the DNA to separate from the excess materials used during the PCR reaction. After that, I worked on my blog.
I had a wonderful Independence Day celebration with my friends on Wednesday and returned to work on Thursday. I felt so excited to receive my first paycheck. When I came to the lab, I made an agarose gel and ran my previous PCR samples on it. It actually worked out well. Afterwards, I made another PCR plate prior to lunch. I made another PCR plate after lunch and I also began labeling my ADHD plates. I then waited for my PCR plates to finish running so that they could be tested on an agarose gel.
Friday was a great day! I began my day by making a PCR plate. I then ran my PCR agarose gel that I made earlier in the week. Fortunately, all my samples worked. Next, I ate lunch early because I was invited to observe a family visit. This is when the proband as well as the entire family come into the clinic area to be tested for ADHD.
Sunday, July 8, 2007
Sequences available!
On Monday, I got the plates I sent off to purification back from the robot on 3rd floor of the Biology building that purifies and sequences plates for the labs. When I used the spec machine to check out my DNA concentration after the purification, nearly all the tubes had a negative concentration! (not good if you want to sequence anything) Unless, of course, the DNA is simply too diluted for the spectrophotometer to read. In that case, there is hope and all is not lost. So I ran a gel on 5 ul of some PCR product comparing it to 1 ul of the stuff I had previously purified myself without using the robot. That way, even if the PCR product was really dilute, I should still be able to see a band. The wierd thing is that all of Lisa's other purifications that were purified along with my samples had no problems with the DNA concentration. At first I thought it might be our spec machine, but I checked some things I had previously purified and they were fine. It's a mystery... So the gel came back with lots of single bands-- meaning my PCR product was in there all along and could be used for sequencing. So I then prepared the plates for sequencing using Big Dye and buffer, but no water since the DNA was already so dilute. That part was easy. When the sequences came back, I got to analyze. What I did on Friday was rename all of the sequences so that their names fit which primer and individual it represents, rather than just a random number. Then I organized the sequences into primer groups. I also got a sneak peek at some SNPs, which were obvious by their characteristic double hump.... more on that next week.
Saturday, July 7, 2007
Mimbulus Mimbletonia
The DNA sequencing results all came back, so for the past two weeks, I've mostly been working on this program called GeneMarker that takes all of the DNA sequencing results and spits out the allele sizes at the EST/microsatellite marker loci as peaks on a graph. Here's a screenshot of a couple of GeneMarker graphs:
 So the peaks portray how many base pairs long different alleles are. However, it's not as easy as it sounds because there are deceitful little peaks that try to trick you into believing that they're real - but really, they're just imposters... Anyway, I've been trying to tackle this program, but it's been putting up a good fight. But I'm determined to conquer it…somehow…
So the peaks portray how many base pairs long different alleles are. However, it's not as easy as it sounds because there are deceitful little peaks that try to trick you into believing that they're real - but really, they're just imposters... Anyway, I've been trying to tackle this program, but it's been putting up a good fight. But I'm determined to conquer it…somehow…
In the greenhouse, I'm still monitoring the germination and flowering of the M. tilingii and M. guttatus; however, many of the tilingii were looking slightly puny, so we ended up replanting some of them.
But thankfully, some of the paired guttatus individuals started flowering, and I began taking down morphological data. We decided on comparing and contrasting flowering time, basal width, overall height, number of vegetative stems, stem thickness between different nodes, internode lengths, leaf length/width, corolla length/width, stigma length, anther length, calyx length/width, and number of flowers after a certain time period.
We're also planning on measuring stomatal density and perhaps (hopefully, if all goes well) trichome density/morphology. Measuring these two traits is really interesting! So for stomatal density, you take casts of the underside of leaves using dental impression material and stick it on a microscope slide. When that dries, you spread some clear nail polish on the casts and make leaf impressions from the nail polish peels. You put those peels on another microscope slide and examine it under a microscope, and you can see individual plant cells, including the guard cells that control stomatal opening/closing! Super cool! Also, I've been wanting to examine more closely the tiny little hair-like structures that are found on both the tilingii and guttatus. Here's an amazing picture of a tilingii individual in which the trichomes are really easy to see (taken by Carrie):
 Amazingly, some of the tilingii populations (like this one) have a sticky substance on their trichomes. It's hypothesized that this glandular exudate is a mechanism for resistance to insect herbivory, insulation, etc. Anyway, we looked at the trichomes on both the tilingii and guttatus and discovered that for the individuals that we examined, the trichome morphology was different between the two species
Amazingly, some of the tilingii populations (like this one) have a sticky substance on their trichomes. It's hypothesized that this glandular exudate is a mechanism for resistance to insect herbivory, insulation, etc. Anyway, we looked at the trichomes on both the tilingii and guttatus and discovered that for the individuals that we examined, the trichome morphology was different between the two species
Oh, and I almost forgot! I was reading Harry Potter and the Order of the Phoenix, and there's this part that describes a plant that Neville has called "Mimbulus mimbletonia". Doesn't "mimbulus" sound eerily similar to "mimulus"? Anyway, I looked it up on Wikipedia:
"Mimbulus mimbletonia is described as "appear[ing] to be a small grey cactus in a pot, except that it was covered with what looked like boils rather than spines" (OotP 186). The genus name mimbulus may be related to the real genus mimulus, especially since those plants are used as folk remedies for "shyness, anxiety, and forgetfulness" and those are traits of Neville Longbottom."Mimulus are definitely not small grey cacti, but I thought that was a cool coincidence. Now whenever I go to the greenhouse, I feel like I'm in Professor Sprout's class! =]
Thursday, July 5, 2007
Romeo oh Romeo....





A post will follow later in the week about my lab week, but I have a few new photos I wanted to put up. Evan showed us this adorable situation. Here are a few pictures of Romeo (diademed sifaka) and the squirrel, a photo of Chloris' baby (ringtail) and one of Eduardo(mongoose). Some of them are a bit fuzzy because I cant use flash.
Monday, July 2, 2007
lab raiding
This week I continued working on my ELISA. I'm trying to optimize the protocol so we can look at the concentration of antibodies in Blood Blank samples. The commercial kits cost $300 each (!). Eventually, the project will look at probably 7500 samples so having an ELISA we can use ourselves makes much more sense. I get to do lots and lots and lots of pipetting. It's always kind of tense for me at the end of the day when I get my results back. To develop the ELISA, I add a substrate for the enzyme attached to the antibodies in the plate. When it develops, it starts to turn blue but often it takes 15-30 minutes to get good results. Sometimes, I just watch the plate as it develops waiting for it to turn blue. After awhile I can't tell if it really is changing color or if it's all in my head hahaha. There are so many places to mess up when I make an ELISA that it's a little stressful waiting for it to turn color. So far they all have though which makes me very happy!
I'm also looking at the subclasses of a particular antibody that patients with HITT have in their plasma. This week I finished optimizing the ELISA for that part so I get to begin collecting data from more samples. That was very exciting because it will *hopefully* get in Dr. Arepally's new paper if the results turn out well.
This week, our lab killed some of the mice they do work with. I didn't go to see because I don't have clearance - I probably wouldnt have gone anyways, it sounds so gruesome. We also raided another lab that was closing so I spent a lot of time carrying stuff back and forth. At one point we have TEN CHAIRS in our tiny lab along with the piles of other stuff we found. It was fun finding all these cool gadgets and chemicals that we needed too.
I'm also looking at the subclasses of a particular antibody that patients with HITT have in their plasma. This week I finished optimizing the ELISA for that part so I get to begin collecting data from more samples. That was very exciting because it will *hopefully* get in Dr. Arepally's new paper if the results turn out well.
This week, our lab killed some of the mice they do work with. I didn't go to see because I don't have clearance - I probably wouldnt have gone anyways, it sounds so gruesome. We also raided another lab that was closing so I spent a lot of time carrying stuff back and forth. At one point we have TEN CHAIRS in our tiny lab along with the piles of other stuff we found. It was fun finding all these cool gadgets and chemicals that we needed too.
Babysitting Jurkat Cells...

Last week was another pretty productive week in the lab. My data study is coming along. A couple of key points are still missing (they're somewhere in our records, but hard to find since they go back to 2006). I also put together data on adult control samples of blood to figure out what a "normal" range is for Natural Killer cells (a type of white blood cell). This is pretty useful since the same adult volunteer can have a big range in NK cells over time, and when you're comparing them to patients it can be misleading.
I also started doing more work in the lab this week. My job was to recover a human T cell line called Jurkat cells. The Jurkat cells needed a specific medium which I put together. Then came "waking them up" which involved finding the right tube in the liquid nitrogen, and heating them quickly in a water bath. The cells are very touchy so you have to be careful and move quickly. I counted the alive cells using a hemocytometer. Every day I checked the cells under a microscope to make sure they were happy and growing. There were a lot of dead cells floating around in the flask but hopefully they don't affect the living ones.
{kind=link}
I also started doing more work in the lab this week. My job was to recover a human T cell line called Jurkat cells. The Jurkat cells needed a specific medium which I put together. Then came "waking them up" which involved finding the right tube in the liquid nitrogen, and heating them quickly in a water bath. The cells are very touchy so you have to be careful and move quickly. I counted the alive cells using a hemocytometer. Every day I checked the cells under a microscope to make sure they were happy and growing. There were a lot of dead cells floating around in the flask but hopefully they don't affect the living ones.
{kind=link}
Later in the week Jie, the same person helping me with the Jurkat cells, gave me a lesson in immunohistochemistry. IHC is just a way to see where different proteins exist in a tissue using antibodies. It was really interesting to see the process, it looked so hard! Each of the slides had different antibodies that would stain the proteins in the tissue in multiple steps. The next day I got to see the slides under a microscope. I was surprised to learn that when a patient gets a thymus transplant the tissue is not put in its natural place, above the heart, but around the thigh where there's more tissue for it to attach to. Under the microscope you could tell the muscle cells from the thymus cells by the shape and by a stain which only colored cells with a thymus markers. We were looking for cells with CMV, a virus, which was almost like finding a needle in a haystack. Looking at control slides of thymus tissue pre-transplantation was also really cool (the pictures above are of normal thymus tissue). Another good week...I can't believe we're almost half way done!
The experiment is working! :-D
I can't believe we're almost half way through these seven weeks! Wow, times goes by really fast. I wish I could have a time machine because I need more time to do my project, haha. Speaking of my project, we finished the first part of it last week. :-D I took measurements of the toluene samples three times a day, and Ruoting took samples to the flow cytometer at UNC to see if horizontal gene transfer between the P. putida and E-coli was occuring. We found that all the bottles, or our "bioreactors," were reacting just as we had expected, except for the ones with E-coli and its conjugate-they weren't degrading toluene. Ruoting and I were very sad. :-( But then, on Saturday, we started a new batch that had lower amounts of toluene, and today, I measured the concentrations, and they went down! That was really exciting. So I'm hoping that the decrease in concentration is due to the E-coli working. Another possibility is that the toluene is escaping from the bottles into the air, but that is very unlikely since they are vaccum sealed. So yay, this week has been really productive, and we might have discovered something! Now I'm going to lunch; I have this McDonald Southern Chicken Sandwhich coupon that I want to use... ;-)
I really liked the AIDS talk that was given last week, and I was shocked that people with salaries over $12,000 couldn't get financial aid for therapies and medicines in North Carolina. Now I have this urge to write a letter to our congressmen about this issue... It'll probably not do any good, but I'll write it anyway. And then we can all sign it! :-)
I really liked the AIDS talk that was given last week, and I was shocked that people with salaries over $12,000 couldn't get financial aid for therapies and medicines in North Carolina. Now I have this urge to write a letter to our congressmen about this issue... It'll probably not do any good, but I'll write it anyway. And then we can all sign it! :-)
Sunday, July 1, 2007
The little urchins are growing!

This week was a pretty interesting one. I'm knee-deep in my project, and the rest of the lab is working on obtaining DNA and other data from millions of baby sea urchins. On Monday I did a HUGE gel for 16 primersets and 12 individuals- that means I loaded 192 wells plus 8 ladders- all within 15 minutes. Beyond that time, the gel begins to get fuzzy and loaded DNA in one well might mix with another well. For this particular gel, I needed some high-tech equipment to fill the wells before the timer ran out. I got to use the collest pipette ever. It was just like a normal automatic pipette except that it had 8 tips instead of 1. This allowed me to load my gel much more quickly and efficiently. it's really fun to use, but hard also. Sometimes the pippette won't suck up or dispense the correct amount of liquid, which can be frustrating. So on Tuesday I got to go to the embryo sea urchin freezer where people were viewing the babies to see if they were developing on schedule. Unfortunately, some of the embryo cultures were infected with bacteria, which had to be eliminated without eliminating the babies. The first thought was to dump the sea water that the eggs were in since the eggs were on the bottom and the water was on top. But some of the eggs had already hatched and the babies were swimming around in the water. Then we used a filter to avoid the eggs and babies, but suction out the bacteria. When I got back to the lab, I looked at the big gel I had run the day before. Turns out one whole plate was ineffective, so I redid that on Wednesday. Thursday I did another gel, and Friday I got to work with the urchins again. By this stage, the urchins look like little floating, transparent pyramids that can swim. They're really cute, but sadly, they had to die for the sake of science... So I spent Friday morning centrifuging the eggs to the bottom of the tube, sucking out the sea water, and replacing it with buffer that instantly kills the larvae and preserves the DNA which is then used to understand more about the offspring. As the little babies grow, they will develop sac-like structures on their sides, from which will spring the future adult sea urchin. The interesting thing about sea urchins is that they are born twice- once from an egg, and then again from these sac-like structures. Why? Nobody knows...
Would risk be different in a female dominated society?
So this week was busy, again, but I had fun in the lab. I created more stimuli, practiced coding and ran more babies.
Something interesting we talked about at lab meeting, which I then continued talking about with Sara and Evan:
Our lab is running risk experiments with both adults, kids and lemurs. The human adults and kids run a gain condition (they risk winning one or three versus two every time) and a loss condition (they lose one every time or risk losing none or two...). The lemurs run only the gain condition. In adults, and I believe in children, boys tend to be more risky. What I talked about with Sara and Evan was the lemurs. Because we don't run any female lemurs, we don't know if this trend would continue. It would be fascinating to see, as female lemurs are dominant (I believe that's what Evan said...). The male ringtails (eulemur catta) tend to run very safe (Aracus often runs 100% safe-two sugar pellets every time). I would love to find out what would happen if we tested female ringtails, however right now none are available.
Something interesting we talked about at lab meeting, which I then continued talking about with Sara and Evan:
Our lab is running risk experiments with both adults, kids and lemurs. The human adults and kids run a gain condition (they risk winning one or three versus two every time) and a loss condition (they lose one every time or risk losing none or two...). The lemurs run only the gain condition. In adults, and I believe in children, boys tend to be more risky. What I talked about with Sara and Evan was the lemurs. Because we don't run any female lemurs, we don't know if this trend would continue. It would be fascinating to see, as female lemurs are dominant (I believe that's what Evan said...). The male ringtails (eulemur catta) tend to run very safe (Aracus often runs 100% safe-two sugar pellets every time). I would love to find out what would happen if we tested female ringtails, however right now none are available.
More of Gas Chambers
Hey Everyone!
This week I continued helping Eileen with her gas chambers. We started our week off by finding the flaws in the chambers. The biggest problem was that the chambers were to hot in the inside. The first time we used them in the field, the plants started to transpire and the inside of the chambers steamed up. So, we spent this week figuring out a more effective cooling method. We spiraled copper tubing around the inside of the chamber and pumped ice cold water through it. Although the water condensed on the tubing and provided the plants with some water, it was the best cooling method we could find. I also got to use the big complicated machinery to drill holes and make the chamber edges sharper once again. Next week we will take the chambers into the field once again, and hopefully we will get the results we want.
This week I continued helping Eileen with her gas chambers. We started our week off by finding the flaws in the chambers. The biggest problem was that the chambers were to hot in the inside. The first time we used them in the field, the plants started to transpire and the inside of the chambers steamed up. So, we spent this week figuring out a more effective cooling method. We spiraled copper tubing around the inside of the chamber and pumped ice cold water through it. Although the water condensed on the tubing and provided the plants with some water, it was the best cooling method we could find. I also got to use the big complicated machinery to drill holes and make the chamber edges sharper once again. Next week we will take the chambers into the field once again, and hopefully we will get the results we want.
Taqman and PCR
Monday was quite interesting. A couple of minutes upon arriving at the Center for Human Genetics Building, the fire alarm went off and everyone had to be evacuated outside the building. When we came back in, I looked at the results for the last for Taqman plates. Sadly, two of the plates failed to amplify, which means that I have to re-do these plates. Then, Chris showed me how to do PCR. He explained the functions of the various ingredients in the master mix. For instance, the taq is an enzyme used for replication and amplification of a DNA fragment. He also showed me how to work the PCR machines. When I finished lunch, I had to re-do the plates that failed. Chris then allowed me to do PCR by myself, which was so exciting. For the remainder of the day, I analyzed the rest of the Taqman plates. In addition, I went into Pub Med and searched for the gene ALAD and lead and neurotoxicity.
Tuesday was a fun day also. I enjoyed the Primate Center tour. I have never seen lemurs in the past and it was amazing to see how closely they resembled humans. In the lab, Chris and I went to use the Biomek robot for transferring DNA into the 384 deep-well plates. At the same time, Robin came to my lab for a lab visit. We talked about various aspects of my project as well as the poster. After lunch, I had a meeting with Dr. Allison and Melanie. We looked at different data and statistics regarding smoking, genotypes, and polymorphisms in relation with ADHD. Amazingly, we found many significance in the data. We also discussed the status of the materials for my project. I then headed back to the lab and did two PCR plates. We actually had a small going away party for two members of the lab who are going to be relocating to Miami. We had some cake and everyone in my lab joined in. Overall, Tuesday went well.
On Wednesday, I was able to make an agarose gel. It was somewhat nerve wrecking to handle even just a small amount of ethidium bromide since it could possibly affect my future children’s DNA. It was, however, interesting how we made a gel with multiple rows of wells rather than just one. The purpose of the gel was to see if our PCRs worked. After running the gel, we took a picture of it and measured the band sizes. Fortunately my first PCRs were great. After lunch, I completed one more PCR plate, and then I proceeded to do two Taqman plates for different SNPs. I then started working on my blogs for thirty minutes. Afterwards, Chris and Dr. Allsion helped me out with my introduction.
On Thursday, I met with Dr. Allison in the morning. She informed me about the upcoming family visit for the ADHD research study patients, wherein they go through a series of tests and get their blood drawn. I am very thrilled to see that. After that, I went to my lab and finished two PCR plates. When lunch was over, I ran the samples from the plates onto the agarose gel to see if my PCR was successful. For the remainder of the day, I completed some more Taqman for patients enrolled in the hostility study.
Finally on Friday, I started planning out the methods section of my poster. Also, I searched about the various SNPs in the genes that I will be studying. I had to find its role and its functions. I made three PCR plates using different primers and ran them on the PCR machine. After that, I made an agarose gel to test my plates on. When lunch was over, I tested my samples from my PCR plates. As it turned out, my PCR turned out well. For the rest of the day, I worked on some more PCR plates and headed to the Schiciano Auditorium for the seminar speaker !! This week was a busy week, but it turned out great!
Tuesday was a fun day also. I enjoyed the Primate Center tour. I have never seen lemurs in the past and it was amazing to see how closely they resembled humans. In the lab, Chris and I went to use the Biomek robot for transferring DNA into the 384 deep-well plates. At the same time, Robin came to my lab for a lab visit. We talked about various aspects of my project as well as the poster. After lunch, I had a meeting with Dr. Allison and Melanie. We looked at different data and statistics regarding smoking, genotypes, and polymorphisms in relation with ADHD. Amazingly, we found many significance in the data. We also discussed the status of the materials for my project. I then headed back to the lab and did two PCR plates. We actually had a small going away party for two members of the lab who are going to be relocating to Miami. We had some cake and everyone in my lab joined in. Overall, Tuesday went well.
On Wednesday, I was able to make an agarose gel. It was somewhat nerve wrecking to handle even just a small amount of ethidium bromide since it could possibly affect my future children’s DNA. It was, however, interesting how we made a gel with multiple rows of wells rather than just one. The purpose of the gel was to see if our PCRs worked. After running the gel, we took a picture of it and measured the band sizes. Fortunately my first PCRs were great. After lunch, I completed one more PCR plate, and then I proceeded to do two Taqman plates for different SNPs. I then started working on my blogs for thirty minutes. Afterwards, Chris and Dr. Allsion helped me out with my introduction.
On Thursday, I met with Dr. Allison in the morning. She informed me about the upcoming family visit for the ADHD research study patients, wherein they go through a series of tests and get their blood drawn. I am very thrilled to see that. After that, I went to my lab and finished two PCR plates. When lunch was over, I ran the samples from the plates onto the agarose gel to see if my PCR was successful. For the remainder of the day, I completed some more Taqman for patients enrolled in the hostility study.
Finally on Friday, I started planning out the methods section of my poster. Also, I searched about the various SNPs in the genes that I will be studying. I had to find its role and its functions. I made three PCR plates using different primers and ran them on the PCR machine. After that, I made an agarose gel to test my plates on. When lunch was over, I tested my samples from my PCR plates. As it turned out, my PCR turned out well. For the rest of the day, I worked on some more PCR plates and headed to the Schiciano Auditorium for the seminar speaker !! This week was a busy week, but it turned out great!
Subscribe to:
Posts (Atom)